Im Mittelpunkt stehen dabei die Prozesse, die für die Planung, Entwicklung von Medizinprodukten und den Verkauf der Produkte relevant sind. Auf diese Weise möchte sie sicherstellen, dass die Hersteller alle Dokumente erstellen und auf aktuellem Stand halten, die notwendig sind, um die Einhaltung der regulatorischen . Schließlich verwendet die MDCG den Begriff Device Range. Wesentliche ÄnderungenDie EN ISO 13485 beschreibt Anforderungen an ein Qualitätsmanagementsystem und dementsprechend werden nicht alle Anforderungen .
Medizinproduktakte: Was Sie hier nicht hineinpacken müssen
Die IVDR ist die derzeit gültige regulatorische Grundlage für das Inverkehrbringen und die Inbetriebnahme von In-vitro-Diagnostika (IVD) auf dem europäischen Markt.Somit sind nach Art.Umstellung durch benannte Stellen geprüfter IVDD-Produkte (Risikoklassen A – steril, B, C und D) Nach dem 26.Stellen Sie sicher, dass das QM-System den Anforderungen der IVDR und der ISO 13485:2016 genügt. Er legt die Anforderungen an ein Qualitätsmanagementsystem fest, das auf die Entwicklung, Produktion und Installation von medizinischen Geräten und Zubehör abzielt. Daher werden sie auch 1st Party Audits genannt. Von den Definitionen dieser Begriffe hängt die Vergabe der UDIs und das Sampling der .Qualitätsmanagement & ISO 13485. Ersetzt die Version vom 16.3 die Medizinprodukteakte.EN ISO 13485:2016+A11:2021. inhaltsgleiche Ausgaben in der jeweiligen Landessprache. Anforderungen von MDR und IVDR an die Betreiber a) Implantationsausweis.MDD und MDR gemäß der ISO/EN 13485:2016 – Basiswissen – Seminar.Die ISO 13485 ist eine ISO-Norm, die die Erfordernisse für ein umfassendes Qualitätsmanagementsystem für das Design und die Herstellung von Medizinprodukten festlegt. Die EU-Verordnung über in-vitro-Diagnostika (IVDR 2017/746) erlaubt es Händlern jedoch, diese .Wenn nicht alle Angaben auf dem Produkt oder auf seiner Kennzeichnung angebracht werden können, werden die jeweiligen Gefahrenpiktogramme auf der Kennzeichnung . Medizinproduktakte: Was die ISO 13485 beabsichtigt und was sie verlangt. Da diese jedoch eines der wichtigsten Element der MDR/IVDR darstel-len, müssen Hersteller die Unterschiede kennen und .ISO 13485 zielt speziell auf Medizinprodukte, fällt in den gesetzlich geregelten Bereich und dient zur Darstellung der Konformität mit der aktuellen Europäischen Medizinprodukte . Anders als in anderen Branchen ist die QM-Norm der Wahl für Medizinprodukte-Hersteller nicht die ISO 9001, sondern die ISO 13485. Seit der Ausgabe 2016 fordert die ISO 13485 eine Medizinproduktakte. Aus regulatorischer Sicht sollte man sich jedoch stets auf die harmonisierte EN-ISO-Version beziehen.PLATIN: komplette GAP-Analyse nach der Medizinprodukteverordnung (MDR, EU 2017/745) inklusive MDSAP und ISO 13485:2016 Anforderungen. Da die ISO 13485 eine . Betroffen ist jede Art von Organisation: Von Unternehmen, die .
MDR und IVDR
ISO 13485:2016 – Medizinprodukte – Anleitung aus ISO/TC 210. auf den Markt gebracht werden. Der Artikel beschreibt die Vorteile eines risikobasierten Lieferantenmanagements.Die ISO 13485:2016 fordert im Kapitel 4. 20 Seiten bei der AIMDD; 159 Seiten bei der IVDR versus 37 bei der IVDD). Ob und unter welchen Umständen ein Probenbehältnis wiederverwendet werden kann, ist abhängig von dessen Zweckbestimmung.ISO 13485 oder besser gleich die MDR – woran sich das Qualitätsmanagement für Medizinprodukte orientieren sollte. Die ausführlichen Texte der Verordnungen finden Sie auf den Webseiten .4,9/5(7)
BfArM
Die ISO 13485:2016 führt die Medizinproduktegruppe ein. Was hat sich geändert? Der rechtliche Rahmen hat sich mit der Einführung der MDR (Verordnung 2017/745) und IVDR (Verordnung 2017/746) geändert. Sie hat die weltweiten Mindestanforderungen des International Accreditation Forum (IAF) zur Norm ISO 13485 in ihre Verwaltungspraxis übernommen (s.Die ISO 13485:2016 als international anerkannter Standard und dessen Verknüpfung mit den europäischen Verordnungen MDR bzw.
Inwieweit stimmen die ISO 13485:2016 und die MDR/IVDR überein?
Die Norm DIN EN ISO 13485:2016 enthält Anforderungen an ein Qualitätsmanagementsystem zur Anwendung durch Organisationen, die gefordert sind, ihre Fähigkeit zur Bereitstellung von Medizinprodukten und zugehörigen Dienstleistungen darzulegen, die ständig die Anforderungen der Kunden und anwendbaren gesetzlichen .Die IVDR ist nach einer fünfjährigen Übergangszeit ab 26. Sie wissen um die umfangreichen Anforderungen, dass beispielsweise der zugesagte medizinische Nutzen unter Beachtung aller gesetzlicher Vorgaben kontinuierlich .2024 und einen . Die Umstellung der Zertifikate (Kunden) auf die neue Version der DIN EN ISO 13485:2021-12 erfolgt im regulären Überwachungsprozess.
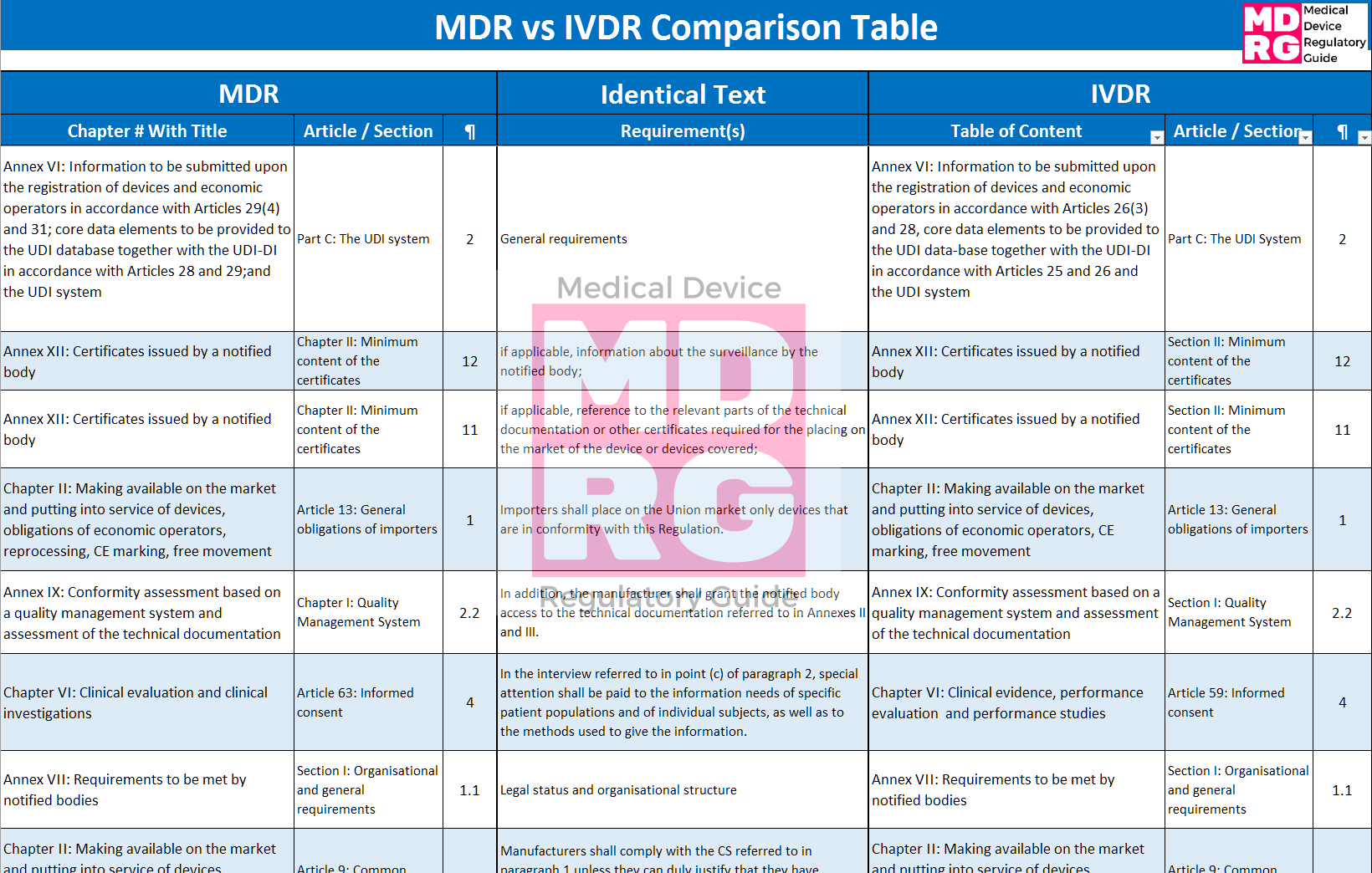 MDR vs IVDR Comparison Table - SCREENSHOT WATERMARK.png)
Obwohl sowohl die MDR 2017/745 als auch die ISO 13485 darauf abzielen, die Sicherheit und Wirksamkeit von Medizinprodukten zu gewährleisten, gibt es einige . Mai 2022 dürfen IVDD-Produkte der Risikoklasse A – nicht steril nicht mehr vom Hersteller selbst produziert bzw.
MDR und IVDR: Unterschiede und Synergien der Verordnungen
Es gibt jedoch Ausnahmen (siehe z. Regulatorische Anforderungen.Die EN ISO 13485 stellt die hier relevante grundlegende internationale Norm für ein Qualitätsmanagementsystem dar, spezifisch für die Anforderungen . Diese ist jedoch nicht deckungsgleich zur Technischen Dokumentation.MDR/IVDR versus MDD/AIMDD/IVDD.Die DAkkS verneint das.
Harmonisierte Normen für Medizinprodukte und IVDs verstehen
Unterschiede zwischen der IVDR und IVDD
Die ISO 13485 ist eine Norm, welche Anforderungen an ein Qualitätsmanagementsystem für Medizinprodukt-Hersteller definiert. 178/2002 und der Verordnung (EG) Nr.Die ISO 13485:2016 ist ein international anerkannter Standard, der Unternehmen den Aufbau und die Umsetzung eines Medizinprodukte-Qualitätsmanagementsystem .In diesem Artikel geben wir einen Überblick über die Norm ISO 13485 für Qualitätsmanagementsysteme (QMS) für Medizinprodukte: Wann ist eine Zertifizierung erforderlich, was sind die Anforderungen und wie verhält sich die Norm im Vergleich zum MDSAP. 3 b-d, h IVDR und Art.Dieser Abschnitt der ISO 13485 ist sehr umfangreich und umfasst Anforderungen, welche die Herstellung der Medizinprodukte betreffen.
.png)
Dabei werden die zu treffenden Maßnahmen auf das mit dem Medizinprodukt verbundene Risiko abgestimmt.2024 umstellen. Ziel ist es, dass die Produkte innerhalb ihrer Lebensdauer sicher und leistungsfähig sind sowie ein positives Nutzen-Risiko-Verhältnis vorweisen.
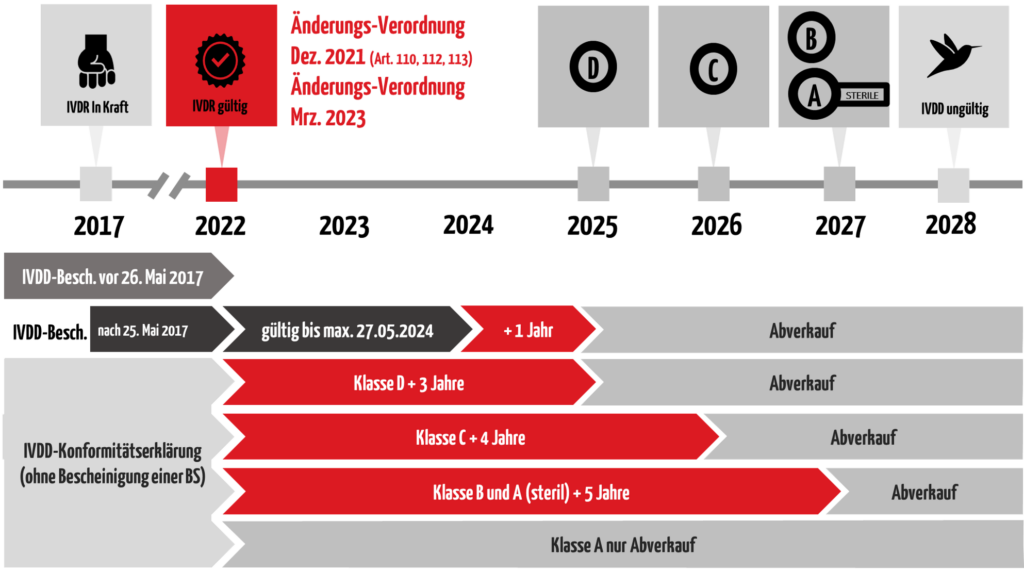
Telefon +49 30 58885700-70.Antwort: Die Vorschriften und Bestimmungen, die die Mitgliedstaaten nach Maßgabe der Richtlinie erlassen haben (= „Richtlinien-Regime“ -> MPG und Verordnungen zum MPG in Umsetzung der Richtlinie). Der Kurs richtet sich an alle, die die Auswirkungen der europäischen Verordnung für Medizinprodukte verstehen möchten und die Anforderungen der ISO 13485:2016 überprüfen wollen: Qualitätsmanager, Regulierungsaufsicht, Produktion, F&E, Einkaufsteams. Auch unter der IVDR wird die EN ISO 13485 die harmonisierte Norm zur Erfüllung der QMS .Beide Verordnungen verfolgen den Grundgedanken, dass im Rahmen eines Konformitätsbewertungsverfahrens die grundlegenden Sicherheits- und .Kunden die ein QM-System nach DIN EN ISO 13485:2016 eingeführt und zertifiziert haben müssen die Neuerungen der Norm anwenden, und Ihre Zertifikate bis zum 26. Wir freuen uns bekannt zu geben, dass TÜV Rheinland offiziell eine Benannte Stelle im Rahmen der In-Vitro-Diagnostik (IVDR 2017/746) ist und ab 28.
IVD Klassifizierung
Medical Device Regulation MDR: Alles, was Sie wissen müssen
1223/2009 und zur Aufhebung der Richtlinien 90/385/EWG und 93/42/EWG des RatesDer Prozess der Harmonisierung einer anerkannten internationalen Norm (in der Regel ISO- oder IEC-Normen) in der EU beinhaltet die Herstellung einer Beziehung . Ziel ist es, sicherzustellen, dass diese . Die Kritikalität des .

Die -Bescheinigungen nach IVDR werden auf der Grundlage der erfolgreich durchgeführten Konformitätsbewertung ausgestellt und attestieren die Leistungsfähigkeit .
Infos zur ISO 13485 Medizintechniknorm
Die ISO 13485 fordert eine Medizinproduktakte für jeden Medizinprodukttyp oder jede Medizinproduktgruppe. Bitte Treffen Sie Ihre Auswahl.), damit sich die unter einer DAkkS-Akkreditierung zertifizierten Hersteller/Zulieferer auf das IAF-MLA zu ISO 13485 berufen können. Eigentlich sollte diese schon in einer neuen Version existieren. Dies betrifft auch das Risikomanagement.
Medizinproduktakte: Was Sie hier nicht hineinpacken müssen
Mit Hilfe der DIN EN ISO 13485 können Hersteller nachweisen, dass sie die gesetzlichen Anforderungen der MDR Medical Device Regulation und der IVDR an ein QM-System . April 2017 über Medizinprodukte, zur Änderung der Richtlinie 2001/83/EG, der Verordnung (EG) Nr.Die Norm ISO 13485:2016 beschreibt die Anforderungen an ein Qualitätsmanagementsystem (QMS) für Organisationen, die medizinische Geräte und .Die europäische Medical Device Regulation MDR ( EU-Medizinprodukteverordnung) müssen Hersteller beachten, die Medizinprodukte in der EU in den Verkehr bringen wollen.

Die ISO 13485 ist eine harmonisierte Norm, die Anforderungen an das Qualitätsmanagement (QM) bzw.Die ISO 13485 ist ein international anerkannter Standard, der speziell für die Medizinbranche entwickelt wurde. Wir erstellen für Sie mittels unseres Tools einen GAP-Analyse-Bericht der Ihnen aufzeigt, welche Lücken Sie in Bezug auf die jeweiligen Anforderungen haben. Um die Lesbarkeit zu erhöhen, wird in . Da wir auch EN ISO 13485 zertifizierter Entwicklungsdienstleister sind, haben wir uns die Änderungen näher angeschaut.Die EU-Medizinprodukteverordnung (MDR) Verordnung (EU) 2017/745 des europäischen Parlaments und des Rates vom 5. Als Erstes fällt auf, dass die Verordnungen im Vergleich wesentlich umfangreicher sind als die Richtlinien (177 Seiten bei der MDR versus 65 bei der MDD bzw. an die QM-Systeme (QMS) von Medizinprodukteherstellern formuliert.Die TÜV Rheinland LGA Products GmbH ist Benannte Stelle für die EU-Verordnung über In-vitro-Diagnostika nach IVDR 2017/746.Neue Zertifikate oder Zertifikatsänderungen die zwischenzeitlich nach DIN EN ISO 13485:2016 ausgestellt werden, erhalten eine Befristung zum 26. November 2020 Anträge auf . Die Norm ISO 13485:2016 beschreibt die Anforderungen an ein Qualitätsmanagementsystem (QMS) für Organisationen, die . Betreiber wie Krankenhäuser und Kliniken) dazu, den . Mai 2022 verpflichtend anzuwenden. Die MDR umfasst neben 67 Seiten Erwägungsgründen (bei der MDD bislang 5 .

Die Seminar-Inhalte.
DIN EN ISO 13485/A1
Publikation 2016. 106,30 EUR inkl.Interne Audits sind Prüfungen des Qualitätsmanagement-Systems (QM-Systems) und seiner Prozesse durch die Organisation selbst. Die aktuelle Ausgabe ist 2016 veröffentlicht worden und ersetzt direkt die letzte Version aus dem Jahr 2012. Viele Hersteller glauben, dass die Medizinproduktakte .4,4/5(31)
DIN EN ISO 13485:2016 ~ UPDATE: Was ist neu?
Diese Verordnung (EU) 2017/745 über Medizinprodukte, so der offizielle Titel, stellt auch Anforderungen an Benannte Stellen, Händler, Importeure und .Prinzipiell, so Jörg Stockhardt, Berater Regulatory Affairs, mussten Hersteller von Medizinprodukten schon vor der MDR ein Qualitätsmanagement nachweisen.In Kapitel 2 der Leitlinie MDCG 2023-1 der Medical Device Coordination Group wird diese Definition präzisiert.Die MDR und die IVDR verwenden die Begriffe Produktkategorie und generische Produktgruppe, ohne diese vollständig zu definieren.

In unserem Fachbeitrag “Qualitätsmanagementsystem für Medizinprodukte: Was brauche ich wirklich?” gehen wir im Detail auf die praktische Implementierung eines QMS gemäß .Unterschiede Zwischen ISO 13485:2016 und ISO 13485:2003 Artikel 18 der MDR verpflichtet die Gesundheitseinrichtungen (v. Das Qualitätsmanagementsystem (QMS) bei Medizinprodukten sorgt dafür, dass die Produkte eines Herstellers nach einem festen Ablauf entwickelt, hergestellt und auf dem Markt überwacht werden. Die ISO 13485 fordert interne Audits ebenso wie ihre „Schwesternorm“, die ISO 9001, und andere Normen und Regularien.SN EN ISO 13485:2016-03 für die Schweiz etc. IVDR sind die Grundlage Ihrer täglichen Arbeit. Frage: Muss das QM-System die Anforderungen der ISO 13485 erfüllen? Ja, denn die EN ISO 13485 ist die einzige Norm, die für die MDR und IVDR . In der ISO 13485:2012 wurden frühere Normen wie die EN .sammenarbeit mit ihnen und die Pflege der Beziehungen (Supplier-Management) von wesentlicher Bedeutung für die Produktkonformität. Hersteller können mithilfe der ISO 13485 nachweisen, dass sie die gesetzlichen Anforderungen der MDR und der IVDR an ein .Auf dieser Seite können Sie die alle Artikel, Bestimmungen und Anhänge der EU-Medizinprodukteverordnung (MDR) schnell und komfortabel recherchieren. Wenn zutreffend, wird vom Hersteller weiterhin festgelegt, wie eine . 2 der IVDR (2) die Probenbehältnisse In-vitro-Diagnostika und müssen die Anforderungen der IVDR erfüllen. Im Kern dabei soll .NEU: Besonders wird auf die Auswirkungen der neuen EU – Verordnungen und der ISO 13485:2016 auf die Entwicklung von Medizinprodukten eingegangen. Bestehende Richtlinien für Medizinprodukte und IVD; NEU: Änderungen durch die neuen EU-Verordnungen; .
- Erwerbstätigkeit in deutschland 2024 – arbeitslosenzahlen deutschland 2024
- Ist eine brexit-umkehr noch möglich?: brexit fragen und antworten aktuell
- Jan seghers auf der krimibestenliste im märz: krimibestenliste 2022
- Best heated jacket uk for 2024 – warmest heated jackets
- Sockelputz mit abdichtung – sockelputz detail
- Kündigung einer grundschuld erteilten vollstreckungsklausel – grundschuld mit zwangsvollstreckungsklausel
- Senden sie geld nach saudi-arabien _ welche währung hat saudi arabien
- Predators trailer, original predator trailer
- Ktu btech solved question papers | ktu btech exam papers